Deconvolution with scaden for spatial transcriptomics data
Compiled: April 12, 2023
Source:vignettes/Deconvolution_with_scaden_for_spatial_transcriptomics_data.Rmd
Deconvolution_with_scaden_for_spatial_transcriptomics_data.RmdRecent studies (Zubair el.al., Wang el.al.) have indicated the potential to apply bulk-deconvolution methods to new technologies such as spatial transcriptomics when the resolution is not at single-cell level.
For this tutorial, we will use scaden to deconvolute a dataset of sagital mouse brain slices generated using the Visium v1 chemistry, and generate cell-type specific gene expression. The data is provided by Seurat package and can be downloaded via InstallData(). If you haven’t installed scaden, please follow instruction here.
Load required libraries and data
## download data if it's not already installed
SeuratData::InstallData("stxBrain")Perform spot deconvolution using scaden
## provide directory that contains python binary to pythonpath, e.g. /usr/bin/
decon_res <- scdecon(bulk = GetAssayData(brain, slot = "data", assay = "SCT"), ref = GetAssayData(refdata_brain,
slot = "data", assay = "SCT"), phenodata = refdata_brain@meta.data, filter_ref = TRUE, decon_method = "scaden",
norm_method = "none", trans_method = "none", marker_strategy = "all", pythonpath = "/path/to/python/bin/dir/",
tmpdir = "/path/to/tmp/dir/")Visualize distribution of cell-type proprotions
prop_barplot(prop = decon_res[[1]], interactive = TRUE)Add predicted cell-type proportions as new assaydata to Seurat object.
prop_assay <- CreateAssayObject(data = decon_res[[1]])
brain[["PROP"]] <- prop_assayVisualize predicted cell proportions for L5 IT, L6 IT and macrophage in spatial
DefaultAssay(brain) <- "PROP"
SpatialFeaturePlot(brain, features = c("L5.IT", "L6.IT"), pt.size.factor = 1.6, ncol = 2)
Compute cell-type specific gene expression
ct_exprs_list <- celltype_expression(bulk = GetAssayData(brain, slot = "data", assay = "SCT"), ref = GetAssayData(refdata_brain,
slot = "data", assay = "SCT"), phenodata = refdata_brain@meta.data, prop = decon_res[[1]], UMI_min = 0,
CELL_MIN_INSTANCE = 1)##
## Astro Endo L2.3.IT L4 L5.IT L5.PT L6.CT
## 20 20 20 20 20 20 20
## L6.IT L6b Lamp5 Macrophage Meis2 NP Oligo
## 20 20 20 20 20 20 20
## Peri Pvalb SMC Serpinf1 Sncg Sst VLMC
## 20 20 20 20 20 20 20
## Vip
## 20
## create assaydata and add to Seurat object
for (i in 1:length(ct_exprs_list)) {
ct_assay <- CreateAssayObject(data = as.matrix(ct_exprs_list[[i]]))
ct_name <- names(ct_exprs_list)[i]
brain[[ct_name]] <- ct_assay
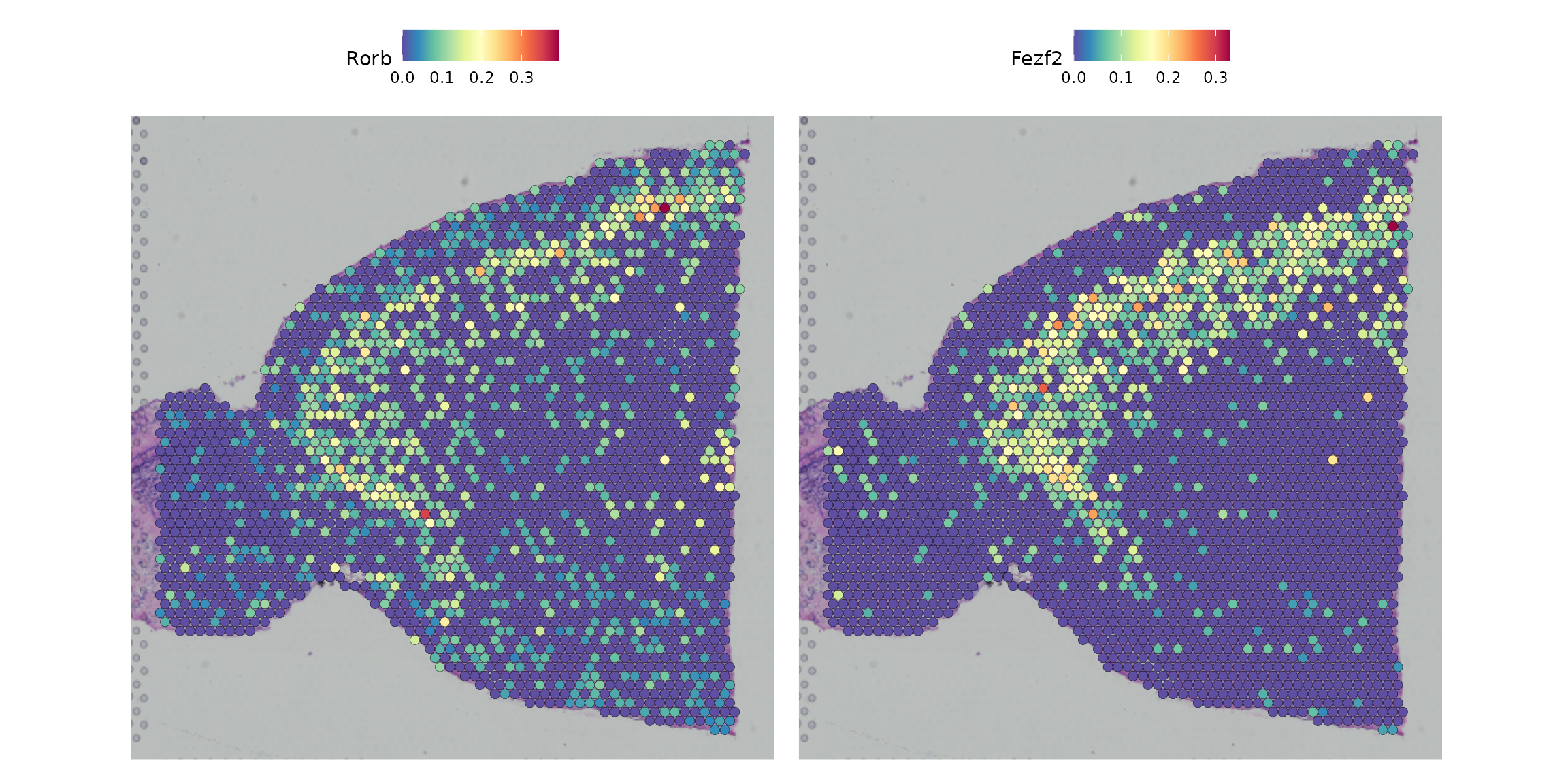
}We can then examine expression of genes using cell-type specific gene expression assays
DefaultAssay(brain) <- "L5.IT"
SpatialFeaturePlot(brain, features = c("Rorb", "Fezf2"), pt.size.factor = 1.6, ncol = 2)